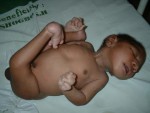
Narození potomka se většinou bere jako radostná událost. Někdy se však při míchání genů nového jedince něco pokazí vysloveně nešťastně a výsledkem je celoživotní těžké postižení, které ovlivní osudy všech členů rodiny. 
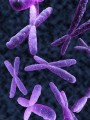
1. Downův syndrom
Příčina: nadbytečný 21. chromozom
Možnost vyléčení: žádná
Délka přežití: 40 let
Četnost výskytu: 1 : 800
Downův syndrom má na svědomí neposlušný 21. chromozom. Ten se odmítá řádně dělit a namísto dvou kopií se do každé buňky vetře hned třikrát. Jeden chromozom navíc není žádná výhra. Nadbytečná várka genů se hádá s ostatními o to, kdo bude plnit jaké povinnosti, a tak se často stane, nakonec se k práci nemá nikdo. Proto často nefunguje velká skupina genů. V případě Downova syndromu se jedná o geny, které mají na starosti vývoj nervových buněk a jejich vzájemnou komunikaci. Absence „výchovných“ genů vadí zejména buňkám šedé mozkové kůry, které ze zoufalství dokonce páchají hromadné sebevraždy. Postižené dítě je proto mentálně i fyzicky retardované a je velmi těžké jej cokoli naučit. Nemocní mívají IQ mezi 30–60.
Nezaměnitelný podpis Downa
K průvodním znakům patří i malá hlava, velká kožní řasa v oblasti očí a oční štěrbiny směřují vzhůru, což zapříčiňuje tzv. mongoloidní vzhled. Hlava je v zadní části zploštělá, posazená na krátký krk s uvolněnou kůží. Končetiny bývají krátké a postižení jsou malé postavy. Děti s Downovým syndromem se často rodí i vývojovou vadou srdce (často dlouhodobě neslučitelnou se životem) a ve zvýšené míře trpí i leukémií.
Varující čísla
V současnosti se pacienti s Downovým syndromem běžně dožívají dospělosti, poslední roky života však tráví v těžké demenci s příznaky podobnými Alzheimerově chorobě. Vadu lze diagnostikovat již v časné fázi vývoje plodu. Možnosti v takovém případě těhotenství přerušit využívá 80 % matek. Četnost výskytu nadbytečného chromozomu se prudce zvyšuje po 30. roku věku. Matkám starším 45 let se takto postižené děti rodí jednou z 25 případů.
2. Těžká kombinovaná imunodeficience
Příčina: poškozené určité geny, většinou umístěné na chromozomu X
Možnost vyléčení: transplantace kostní dřeně, výhledově genová terapie
Délka přežití: 20 let
Četnost výskytu: 1 : 100 000
Vlivem zpřeházených písmen v genetických plánech se nevyrábějí protilátky a nevznikají bílé krvinky. Tělu tak chybí jakákoli obrana proti veškerým infekcím. Stačí pár neurvalých částeček obyčejného chřipkového viru a takto postižené dítě zaplaví smrtelná infekce. Organismus dokonce nepřežije ani očkování živými vakcínami, na které zdravé děti reagují jen lehce zvýšenou teplotou a tvorbou protilátek. Dát si dobré jídlo v restauraci nebo si jen třeba vyjít do lesa je pro takové děti nesplnitelný sen.
Smrtící naděje
Bez složité léčby a celoživotního pobytu ve sterilním prostředí se pacienti dožijí jen několika měsíců věku. V současnosti existuje možnost transplantace zdravé kostní dřeně, která dokáže vyrábět bílé krvinky a čelit tak nájezdům nepřátelských mikroorganismů. Často však není k dispozici vhodný dárce a samotná transplantace může přinést smrtelné komplikace. V 90. letech se začala používat genová terapie. Vědci připravili zdravou variantu genů, které měl paciente poškozeny. Ty pak přibalili do výbavy spřátelenému viru a vypustili jej do postiženého organismu. Jeho úkolem bylo zdravé geny vpašovat do DNA. To také udělal a několik měsíců se zdálo, že vše funguje přesně tak, jak si vědci naplánovali.
Nešikovný agent
Pak ale přišla studená sprcha. Děti začaly po genových terapiích trpět leukémií. Ukázalo se, že virus sice poslušně nastrkal geny do pacientovy DNA, ale udělal to tak nešikovně, že zablokoval dohled nad množením buněk. Ty pak začaly nekontrolovatelně leukemicky bujet. Proto je v současnosti genová terapie za pomoci virů zakázána a čeká se na nový vhodný přenašeč.
3. Syndrom Holterové-Oramův
Příčina: dominantní verze genu TBX5
Možnost vyléčení: žádná
Délka přežití: běžný věk
Četnost výskytu: 1 : 100 000
Jedná se o jednu z mála těžkých dědičných vad, za kterou může „agresivní“ projev jednoho jediného genu. Pokud člověk má tu smůlu, že dostane do výbavy dominantní variantu genu nazvaného TBX5 (bohatě stačí jedna), syndrom se u něj pokaždé projeví. To ovšem představuje i výhodu v tom, že zdraví rodiče se nemusejí obávat výskytu onemocnění u svých potomků. Nová mutace, která neškodnou recesivní variantu změní na dominantní TBX5, se vyskytuje jen velmi vzácně.
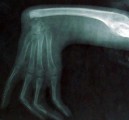
Nepovedené kosti a poškozené srdce
Nemoc se projevuje tím, že při vývoji plodu poplete vytváření kostí minimálně jedné horní končetiny. Dítě se pak rodí bez ruky, případně s rukou zakrnělou, s nedostatečně vyvinutými kostmi nebo zcela bez kostí. Někdy jsou končetiny zdánlivě v pořádku, nicméně na rentgenu je patrné, že kosti nejsou na svých místech. U 75 % pacientů se objevují i závažné srdeční problémy, kdy se zcela neoddělí pravá a levá polovina a komory od síní.
Sedativum hrůzy
V 50. a 60. letech minulého století se na světě narodilo desetitisíce dětí s podobnou vadou, jakou způsobuje TBX5. Tenkrát však za jejich nemocí stálo špatně otestované německé sedativum thalidomid. To mělo za následek, že zcela zastavilo vývoj tkání podílejících se na tvorbě určité části končetiny. Lékaři je po dobrých výsledcích na hlodavcích a psech předepisovali i těhotným ženám. Než se odhalila spojitost mezi deformovanými končetinami a užíváním thalidomidu, trvalo to 6 let. Na základě těchto událostí se stanovila nová přísná pravidla pro testování léčiv.
4. Duchenneova svalová dystrofie
Příčina: mutace genu DMD na pohlavním chromozomu X
Možnost vyléčení: žádná, výhledově genová terapie
Délka přežití: 20 let
Četnost výskytu: 1 : 3500 (u chlapců)
Na chromozomu X za běžných okolností sedí gen, který má na starosti správný vývoj buněčných obalů svalových buněk. Když se porouchá, svaly se nechávají utlačovat od okolních buněk a jejich hmotu postupně nahrazuje tuková a pojivová tkáň.
Dívky ve výhodě
Vzhledem k tomu, že se gen nachází na pohlavním chromozomu X, mají chlapci k dispozici jen jeho jednu kopii (muži mají jeden pohlavní chromozom X a jeden Y). Na chromozomu Y nic podobného k mání není. Choroba se tedy u nich vždy projeví v plné síle. Oproti tomu dívky dostávají do výbavy dva chromozomy X, a tedy i dvě kopie tohoto genu. Když se jedna z nich porouchá, může druhá zdravá varianta zvládnout práci sama. V takovém případě nositelky nepozorují žádné příznaky. Přesto často bývají průvodními znaky vývojové vady srdce. Ale i dívkám se může stát, že jejich buňky odstaví zdravý chromozom a berou si informace pouze z toho nemocného. Pak u nich choroba projeví stejně jako u chlapců.
Plíživá smrt
Nefunkční svaly se velmi výrazně podepisují na schopnosti vykonávat jakýkoli pohyb. Potíže nejsou znatelné hned po narození, ale začínají se projevovat kolem 3.–5. roku věku podivnou chůzí. Po 5. roce se dostavují potíže se vstáváním a do 12. roku je většina pacientů odkázána na invalidní vozík, dochází k téměř naprostému ochrnutí. Do dospělosti většina z nich umírá na dýchací potíže a srdeční vady.
5. Cystická fibróza
Příčina: mutace genu CFTR
Možnost vyléčení: žádná, výhledově genová terapie
Délka přežití: 40 let
Četnost výskytu: 1 : 3000
Nemoc se projeví pouze v případě, že se v jednom organismu sejdou dva poškozené geny zvané CFTR, jejichž nepravosti nedává do pořádku žádná zdravá verze. Je tedy zapotřebí, aby oba rodiče byli přenašeči skryté vlohy pro toto onemocnění.
Špatně namazané trubky
Cystickou fibrózu způsobuje mutace genu, ve kterém jsou zapsány správné postupy na výrobu hlenu pokrývajícího sliznice vnitřních orgánů. Tento hlen je jakousi ochrannou vazelínou, která brání poranění, vyschnutí a infekci citlivých tkání. Pokud gen CFTR funguje špatně, je hlen příliš hustý (přibližně desetkrát hustší než u zdravého člověka), hromadí se v dýchacích nebo trávicích cestách a ucpává je. Nánosy hlenu jsou také živnou půdou pro různé mikroorganismy, které si tu dopřávají orgie a vytvářejí záněty.
Utrápené plíce
Výsledkem je, že postižený člověk má problémy s dýcháním, a trpí častými plicními infekcemi. To vede k postupnému nahrazování funkční tkáně vazivem a plíce tak ztrácejí svou funkci. Pacienti trpí podobným problémem i u slinivky. Tam hustý hlen zabraňuje vypouštění trávicích šťáv do střev. Přijímaná potrava tedy nemůže být dobře zpracována a pacienti trpí různými metabolickými poruchami.
Vražedné řádění chromozomů
Downův syndrom je nejčastější ze tří poruch zapříčiněných nadbytečným nepohlavním chromozomem, které jsou ještě slučitelné se životem. Také u zbývajících dvou se riziko výskytu značně zvyšuje s věkem matky.
Edwardsův syndrom (tedy přebytečný 18. chromozom) se kromě těžké mentální retardace projevuje i deformací končetin. Dítě přežívá jen několik měsíců. Četnost výskytu je u živě narozených dětí desetkrát nižší než u Downova syndromu (1 : 7500). To je ovšem dáno vysokou úmrtností v prenatálním období, kdy tuto vadu nepřežije 95 % zárodků.
V případě, že se v jádře vyskytne 13. chromozom ve třech kopiích, jedná se o Pataův syndrom. Ten má na svědomí těžkou fyzickou i mentální retardaci, zmenšený mozek, často velké mezery mezi lebečními kostmi, špatně vyvinuté nebo zcela chybějící oči. Častými průvodními znaky bývá i rozštěp rtu a patra a zvýšený počet prstů na končetinách. Deformované bývají i vnitřní orgány, nejčastěji srdce, ledviny a pohlavní soustava. Vada se vyskytuje u jednoho z 25 000 živě narozených dětí. Postižení se dožívají maximálně několika měsíců.
Středověcí mutovaní baviči
Jedna z dědičných chorob, která má velmi negativní vliv na zevnějšek pacientů, s sebou přináší i určitou výhodu. Tzv. achondroplazie je způsobená mutací v jediném genu. Ta zařídí špatný růst kostí a pacientovi zkrátí končetiny i trup, a naopak ponechá větší hlavu. Inteligence takto postižených lidí bývá nadprůměrná. Postižení často svůj úděl zvládají za vydatné pomoci notné dávky smyslu pro osobitý humor. Proto se také ve středověku často stávali společníky mocných jako obveselující šašci, kteří bývali i skrytými rádci. Pro jejich břitký úsudek a intrikářské schopnosti bývali obávanými postavami královských dvorů.